
分子動力学ソフトLAMMPSを用いたミセル自己形成のシミュレーション
Last Update:2021/12/09
1. はじめに
分子動力学ソフトLAMMPSを用いて、ソフトマターの問題を取り扱うことが可能です。特に膜などの構造が形成される様子をシミュレーションして可視化すると、印象的な動画を比較的簡単に作成することができます。ここではLAMMPSのパッケージに含まれるmicelleという例をとりあげて、可視化ソフトovitoで可視化してみます。動作環境はMateriApps LIVE! version 3.3を用いています。
2. 実行準備
まずホームディレクトリに作業ディレクトリを作り、作業ディレクトリへ移動します。
cd $HOME mkdir lammps cd lammps
次にLAMMPSのexamples/micelleディレクトリをコピーし、そこに移動します。
cp -r /usr/share/lammps/examples/micelle ./ cd micelle
3. 入力ファイルの編集
micelleディレクトリに入力ファイルin.micelleがあります。計算途中の状態を可視化するために、このファイルを編集します。
emacs in.micelle
編集ソフトは何でも構いません。ここではemacsを使っていますが、MateriApps LIVE!にはvim, nano, Mousepadなどのエディタがはいっていますので、好みのエディタを使ってください。ファイルの内容は以下のようになっています。
# 2d micelle simulation dimension 2 neighbor 0.3 bin neigh_modify delay 5 atom_style bond # Soft potential push-off read_data data.micelle special_bonds fene ...(中略)... thermo 50 #dump 1 all atom 2000 dump.micelle ...(中略)... reset_timestep 0 run 1000
はじめのところで、dimension 2によって、二次元の計算を指定しています。ファイルの最後の方にある#dump 1から始まる行について、最初の#をとって、以下にようにします。
dump 1 all atom 2000 dump.micelle
さらに入力ファイルの最後の行にあるrun 1000という行を以下のように書き換えます。
run 100000
書き換えが終わったらファイルをセーブしてエディタを終了させます。(Emacsであれば、ctrl-x ctrl-sでセーブ、ctrl-x ctrl-cで終了になります。)
4. LAMMPSによる計算の実行
編集した入力ファイルin.micelleを使って、LAMMPSを実行します。
lammps < in.micelle
エラーメッセージなしで、最後にTotal wall timeが表示されていれば、無事実行が完了しています。このとき同じディレクトリにdump.micelleというファイルが生成されています。(lsコマンドで確認できます。)
5. ovitoによる可視化
生成されたファイルをovitoで開きます。
ovito dump.micelle
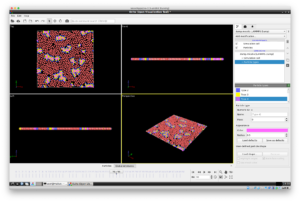
以下のようなスクリーンショットが現れるはずです(以下は再生ボタンを押したあとの画面ですが、ミセルがまだ形成されていない画面がでるはずです)。右下のボタンを押せば、動画が再生されます。粒子が集まってミセル構造を形成する様子をみてとることができます。(見やすくするために、画面中のParticle typesから各粒子の表示半径を0.5に変更してあります。)
6. おわりに
比較的小規模の分子動力学計算であれば、このように簡単に可視化が可能です。多くの粒子を含む系を可視化をする場合は、MateriApps LIVE!で行うと処理が重くなりますので、通常のデスクトップ上に別の可視化ソフト(VMDなど)をインストールし、そこにデータを移行させて可視化するほうが効率がよくなります。