Quantum ESPRESSOと物質データベースを利用した電子状態計算
Last Update:2021/12/09
1. はじめに
Quantum ESPRESSO (QE)を用いて第一原理計算を行うには、結晶構造について事前に情報を取得しておく必要があります。すでにGaAsの第一原理計算について紹介していますが、一般に物質の構造は複雑であり、一から準備するのは面倒です。ここでは物質データベースから結晶構造のデータを取得し、その結晶構造に対して第一原理計算を実行する方法を説明しましょう。物質としては、遷移金属ダイカルコゲナイドの一つであるWSe2を取り上げてみよう。もともとは3次元物質ですが、層状構造を持っており、スコッチテープなどを使って剥がすことで単層のWSe2を取り出すことができます。ここでは単層のWSe2のバンド計算を行ってみましょう。利用したMateriApps LIVE!のバージョンは3.3, Quantum ESPRESSOのバージョンは6.7です。
2. 結晶構造データの取得
結晶構造データベースにはいくつかの種類がありますが、ここではMaterias Projectを利用しましょう。MateriApps LIVE!上でブラウザを立ち上げます。ブラウザはスクリーン左下のスタートボタンの2つ右側の地球儀のボタンを押すと立ち上がります。立ち上がったブラウザでMaterials Projectを検索すれば、すぐにMaterials Projectのページに辿り着くことができるでしょう。まず登録が必要となりますが、トップページからSign in or Resigterのボタンを押して指示に従って登録することができます。googleもしくはgithubアカウントを持っているとすぐに登録できます。メールによる登録も可能です。すぐにログインできるようになるはずです。
登録が終わったらMaterials Projectにログインしてみましょう。周期律表が表示されるはずです。周期律表のWとSeのボタンをクリックしてから、Searchボタンを押してみましょう。様々な構造が表示されますが、単層のWSe2の結晶構造はリストの一番下のmp-1023936とかかれた構造です。(結晶構造は通常いくつか表示されますが、結晶群やユニットセル当たりの原子数などがかかれていますので、それを参考にして選択を行います。)この欄をクリックすると、結晶構造が表示されます(結晶構造はマウスを使って回転させることができます)。ちなみに下のほうにはバンド構造も表示されています。今から行う計算が正しいかどうかは、ここに示されたバンド図と比較すればすぐにわかります。結晶構造が表示されている欄の下にCIFというボタンがあり、これをクリックしたあと、Primitive Cellを選択し、新しく開くダイアログボックスでSaveを選択し、OKボタンを押しと、結晶構造ファイル形式(.cif)のダウンロードが開始されます。ダウンロード先はデフォルトではホームディレクトリ下のDownloadsフォルダになっています。
実際に正しく結晶構造ファイルがダウンロードされているか、VESTAを使って確かめてみましょう。画面左下のスタートメニューからEducation→VESTAを選択し、VESTAを起動させます。File→Openを選択し、Downloadsフォルダを指定すると、さきほどダウンロードしたファイルがあるので選択してOpenボタンを押します。下のような結晶構造が表示されればOKです。ここでも結晶構造はマウスで回転させることができます。

3. Quantum ESPRESSO用の入力ファイルの生成
ダウンロードした結晶構造ファイルから、Quantum ESPRESSO用の入力ファイルを生成してみましょう。そのためにはcif2cellというツールを用います。ダウンロードしたファイルをコピーしてきて、cif2cellによって変換してみましょう。
cp ~/Downloads/*.cif . cif2cell -p pwscf -f WSe2_mp-1023936_primitive.cif -o WSe2.template.in
書式はcif2cell -p (プログラム名) -f (cifファイル名) -o (出力ファイル名)です。実際にできたファイルを眺めてみましょう。
cat WSe2.template.in
最初にコメントがあり、コメント以下は次のような内容になっていることがわかります。
&SYSTEM
ibrav = 0
A = 3.32561
nat = 3
ntyp = 2
/
CELL_PARAMETERS {alat}
1.000000000000000 0.000000000000000 0.000000000000000
-0.500000000000000 0.866025403784439 0.000000000000000
0.000000000000000 0.000000000000000 5.270333379074286
ATOMIC SPECIES
Se 78.96000 Se_PSEUDO
W 183.84000 W_PSEUDO
ATOMIC POSITIONS {crystal}
W 0.000000000000000 0.000000000000000 0.000000000000000
Se 0.333333333333333 0.666666666666667 0.095876000000000
Se 0.333333333333333 0.666666666666667 0.904124000000000
このようにして原子位置の情報は得られましたが、ATOMIC SPECIESの部分で擬ポテンシャルの指定が抜けていますし、そのほかの計算条件の部分はユーザがエディタなどを用いて加筆しなければいけません。
4. 入力ファイルの準備と実行
WSe.template.inから加筆を行ったもののサンプルを下記のように取得します。
wget https://github.com/cmsi/malive-tutorial/releases/download/tutorial-20200514/qe_WSe2.tgz
ダウンロードしたファイルを解凍し、できたディレクトリに入ります。
tar zxvf qe_WSe2.tgz cd qe_WSe2
ここに入力ファイルがそろっています。まずcat WSe2.scf.inを実行して、ファイルの中身をみてみましょう。コメント行以下は次のようになっています。
&control
calculation = 'scf'
prefix = 'WSe2'
pseudo_dir = './'
wf_collect = .true.
/
&SYSTEM
ibrav = 0
A = 3.32561
nat = 3
ntyp = 2
ecutwfc = 50
ecutrho = 400
/
&electrons
mixing_mode = 'plain'
mixing_beta = 0.7
conv_thr = 1.0d-8
/
CELL_PARAMETERS {alat}
1.000000000000000 0.000000000000000 0.000000000000000
-0.500000000000000 0.866025403784439 0.000000000000000
0.000000000000000 0.000000000000000 5.270333379074286
ATOMIC_SPECIES
Se 78.96000 Se.pbe-dn-kjpaw_psl.1.0.0.UPF
W 183.84000 W.pbe-spn-kjpaw_psl.1.0.0.UPF
ATOMIC_POSITIONS {crystal}
W 0.000000000000000 0.000000000000000 0.000000000000000
Se 0.333333333333333 0.666666666666667 0.095876000000000
Se 0.333333333333333 0.666666666666667 0.904124000000000
K_POINTS {automatic}
6 6 1 0 0 0
必要なデータが加筆されているのがわかると思います。それぞれのパラメータの意味はこのレビューを参考にしてください。次に必要は擬ポテンシャルをダウンロードしましょう。ブラウザを立ち上げて、Quantum ESPRESSO pseudopotentialsのページに入ります(quantum espresso pseudoで検索するとすぐにURLがわかります)。トップページの周期律表からWを選択してみてください。様々な種類の擬ポテンシャルが表示されますが、ここでは一番上の擬ポテンシャル(W.pbe-spn-kjpaw_psl.1.0.0.UPF)をつかいましょう。一番上をクリックするとファイルの中身が表示されます。ここでブラウザの右上の三本横線マークをクリックし、Save Page Asを選択して、ページをセーブします。同様にして、Seも一番上の擬ポテンシャル(Se.pbe-dn-kjpaw_psl.1.0.0.UPF)をダウンロードしておきましょう。
ダウンロードしたファイルはDownloadsフォルダにあるので、これをコピーしてきます。
cp ~/Downloads/*.UPF .
これで準備完了です。バンド計算を実行してみましょう。以下のコマンドを実行します。(それぞれ数分程度かかります。)
pw.x < WSe2.scf.in > WSe2.scf.out pw.x < WSe2.nscf.in > WSe2.nscf.out bands.x < WSe2.band.in > WSe2.band.out
無事終了したら、下記のコマンドを実行してみましょう。
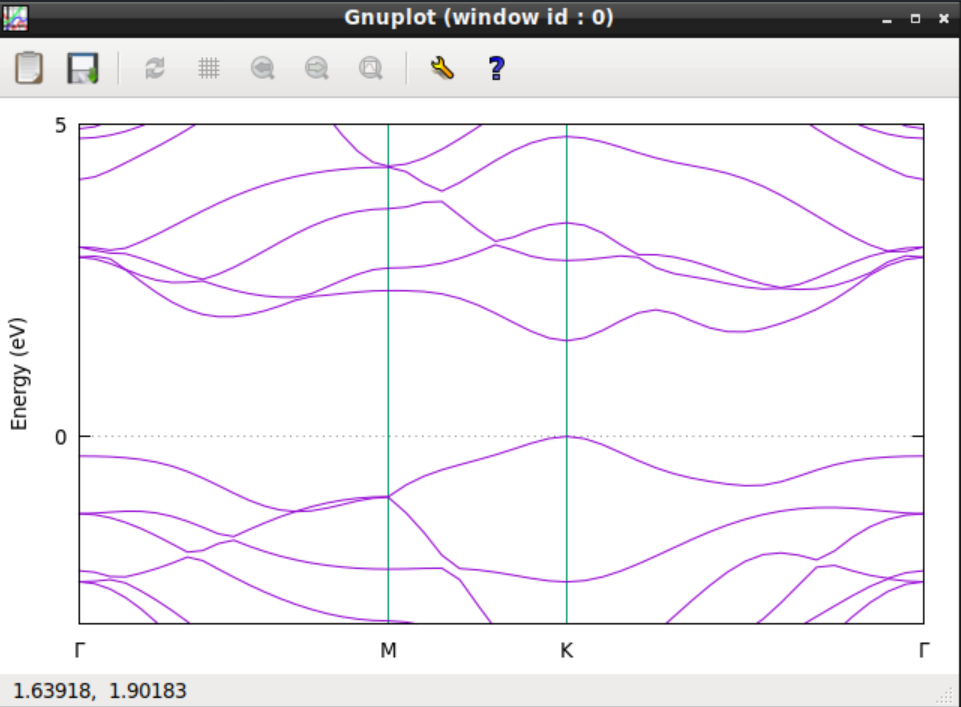
gnuplot -persisent plotWSe2.gp
バンド図が表示されたら成功です。またこのバンド図をMaterials Projectにかかれているバンド図と比較してみましょう(ここではバンド図の一部だけ計算していますので、Materials Projectのバンド図の左半分と対応しています)。バンドギャップがおよび1.5eVであることがわかります。ただし、バンド計算ではバンドギャップの大きさは正確には再現しないことに注意してください。