
LAMMPSを用いたアルミニウムの応力歪み曲線の計算
Last Update:2021/12/09
はじめに
LAMMPSは代表的なオープンソースの分子動力学ソフトウェアの一つであり, 固体やソフトマターなどの物性評価に広く用いられている. 原子・分子だけでなく, レナード・ジョーンズ型の相互作用をもつ粒子系や連続体模型から得られる有効粒子模型など, より広いクラスの物理系にも適用可能である. LAMMPSではソフトウェア内に標準的な力場が多数用意されている. LAMMPSの公式Webページには様々な情報が提供されているほか, 様々な解説・チュートリアルがWeb上で公開されており, 初心者が始めやすい環境が整っている. ここでは, MateriApps LIVE!上でLAMMPSを用いてアルミニウムの応力-歪み曲線を計算してみよう。
本チュートリアルで使用したMateriApps LIVE!のバージョンは2.4である.
MateriApps LIVE!上での実行方法
MateriApps LIVE!を起動するまえに、「設定>システム>プロセッサー」でVirtualBox内で使用するCPUプロセッサの数を可能な限り増やしておくとよい(PCのCPUコア数を調べ、それと同じにしておくとよい. 通常はCPU数選択画面で緑のバーの範囲内で最大に設定すればよいはずである.). 並列計算を行ったほうがより短い時間で計算を終了することができる. MateriApps LIVE!を起動・ログインしたら, まずは画面左下のスタートメニューから「System Tools>LXTerminal」を選択して端末を立ち上げる.ここから, MateriApps LIVE!用に準備されたスクリプトファイルをダウンロードして実行してみよう(このスクリプトファイルでは, Web上のチュートリアルに基づいており, アルミニウムの一軸圧縮を行ったときの圧力を計算する. 詳細情報はチュートリアルを参照していただきたい). このスクリプトをダウンロード・実行するには, 端末で以下のコマンドをタイプし, 1行ごとに改行キーを押す:(以下、行頭の$ は端末のプロンプトを示している)
$ wget https://github.com/cmsi/malive-tutorial/releases/download/em2020/lammps.tgz $ tar zxvf lammps.tgz $ cd lammps $ mpirun -np [ 並列数 ] lammps -in in.comp.txt $ gnuplot -persistent plot.gp
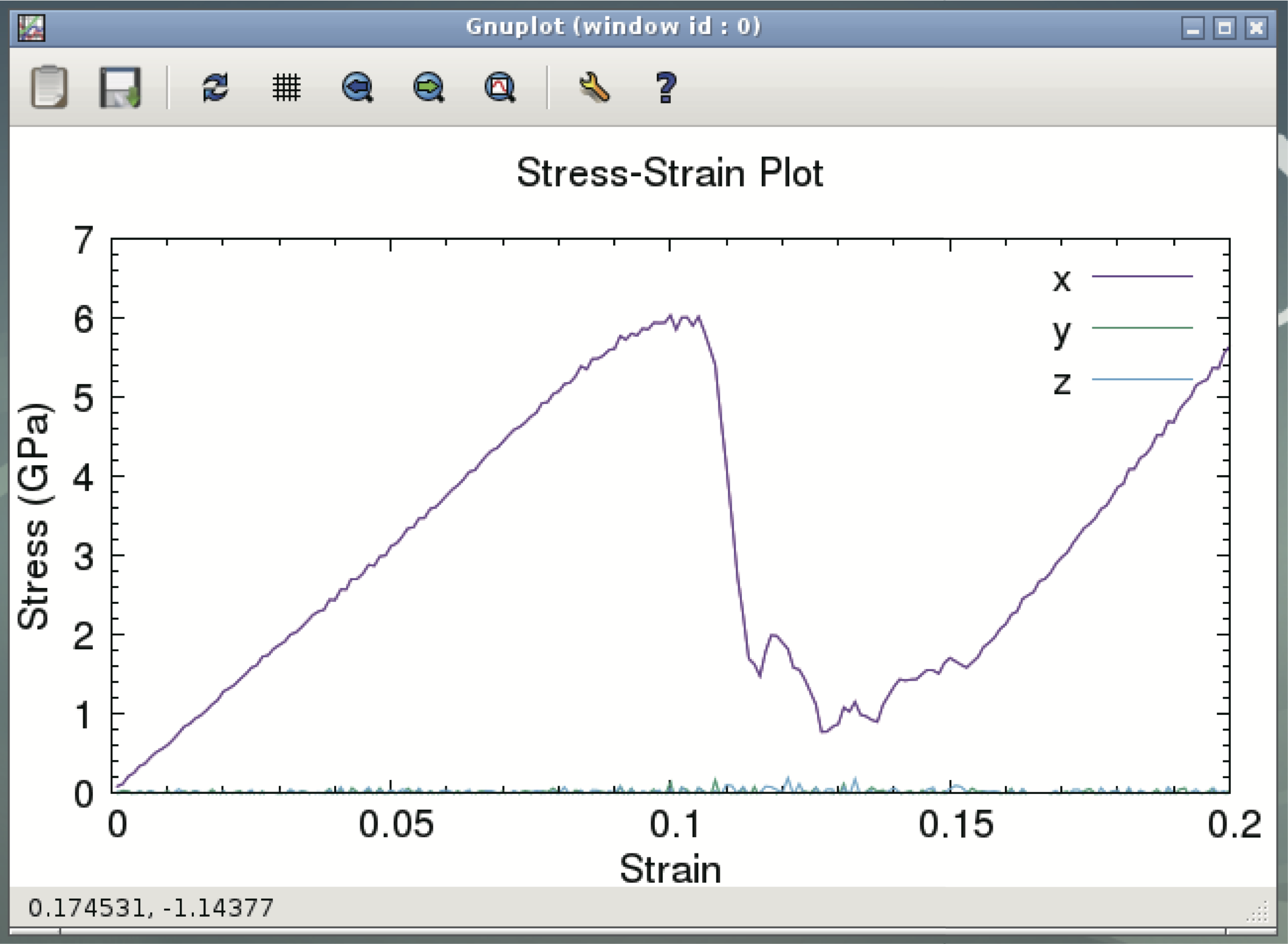
最後から二番目のコマンドでLAMMPSによる分子動力学計算を実行する. [並列数]と書かれた部分にはVirtualBoxで設定したCPU数を書く. 指定する並列度(=利用するCPUの数)にもよるが, 通常はノートパソコンでも10分以内には終了するはずである. 最後のコマンドでアルミニウムの応力-歪み曲線(下図)が表示される. ある歪み以上で応力が急速に変化しているのは, 系に転位などの大きな変形が導入されるからである.
入力ファイルの概要
LAMMPSでの計算に必要となる入力ファイル(上述の例ではin.comp.txt)の中身を少し見てみよう(カギカッコ内は筆者のコメント):
# INITIALIZATION
units metal [ 単位系の指定 ]
dimension 3 [ 扱う系の次元 ]
boundary p p p [ 周期境界条件 ]
atom_style atomic [ 粒子の種類 ]
variable latparam equal 4.05 [ 格子間隔 ]
# ATOM DEFINITION
lattice fcc ${latparam} [ 面心立方格子 ]
region whole block 0 10 0 10 0 10 [ 単位格子数 ]
[ ・・・中略・・・ ]
# FORCE FIELDS
pair_style eam/alloy [ 原子間ポテンシャルの種類 ]
pair_coeff * * Al99.eam.alloy Al [ 力場の指定 ]
[ ・・・後略・・・ ]
単位の指定をmetal(2行目)にしたときは, 長さの単位はÅである. このように扱う系の原子配置や境界条件, 力場の指定を入力ファイルに記述することで, 分子動力学計算の対象や計算条件を細かく指定することができる(詳しくはLAMMPSのドキュメントを参照). 最後の2行では力場の指定を行っている. 力場の選択は分子動力学計算を行う上で最も重要であり, 考える系に応じて適切に選ぶ必要がある(研究に用いる場合には, 力場モデルの詳細について原著論文をよく調べ, 力場モデルの性質や精度, 適用限界などについてよく把握すべきであろう. ). 例えば, 上記の例ではEmbedded Atom Method(EAM法, 原子間の2体相互作用のほかに, 着目する原子の場所に他の原子がつくる平均ポテンシャルが付加された模型)を使っている.LAMMPSの中に用意されている力場セットのほかに, データベースから力場の情報をダウンロードして使用することも可能である.
おわりに
LAMMPSには設定項目が多く、使い始めはとっつきにくいかもしれない。しかし材料分野で様々な応用事例が公開されており、自分の計算したい系にあった計算方法・入力ファイルの例をWebで見つけることはそれほど難しくない。使い慣れれば、非常に強力な分子動力学アプリである。ぜひ使ってみていただきたい。