
Calculation of the stress-strain curve of aluminum using LAMMPS
Last Update:2021/12/09
Introduction
LAMMPS is one of the representative open-source software for molecular dynamics, and is widely used for evaluating physical properties of solids and soft matter. Not only atoms and molecules but also particles with the Leonard-Jones type interaction can be treated. It is also applicable to a wider class of physical systems, such as the effective particle model obtained from continuum models. LAMMPS provides a number of standard force fields in the software. In addition to providing various information, tutorials are published on the Web, providing an environment that is easy for beginners to get started. Here, let’s calculate stress of aluminum under uniaxial pressure by using LAMMPS on MateriApps LIVE!
The version of MateriApps LIVE! used in this tutorial is 2.4.
How to run on MateriApps LIVE!
Before starting MateriApps LIVE !, increase the number of CPU processors used in VirtualBox as much as possible in “Settings > System > Processors” (Normally, it should be set to the maximum value within the range of the green bar in the CPU number selection screen). Of course, the parallel computation make s calculation time shorter. After launching MateriApps LIVE! and logging in, first select “System Tools > LXTerminal” from the start menu at the bottom left of the screen to start up the terminal. Download and execute the script file prepared for MateriApps LIVE! (This script file is based on a tutorial on the web, and calculates the pressure of aluminum under uniaxial compression. For more information, see the tutorial. To download and run this script, type the following commands in the terminal and press the Return key after each line: (The $ at the beginning of the line is the terminal prompt.) Is shown)
$ wget https://github.com/cmsi/malive-tutorial/releases/download/em2020/lammps.tgz $ tar zxvf lammps.tgz $ cd lammps $ mpirun -np [ parallel number ] lammps -in in.comp.txt $ gnuplot -persistent plot.gp
Execute the molecular dynamics calculation by LAMMPS with the second line from the end of the above command. Write the number of CPUs set in VirtualBox in the part where [parallel number] is written. Standard laptop computers usually finish calculation within 10 minutes. The last command displays the aluminum stress-strain curve (see below). The stress changes rapidly above a certain strain. This is because large deformations such as dislocations are introduced into the system.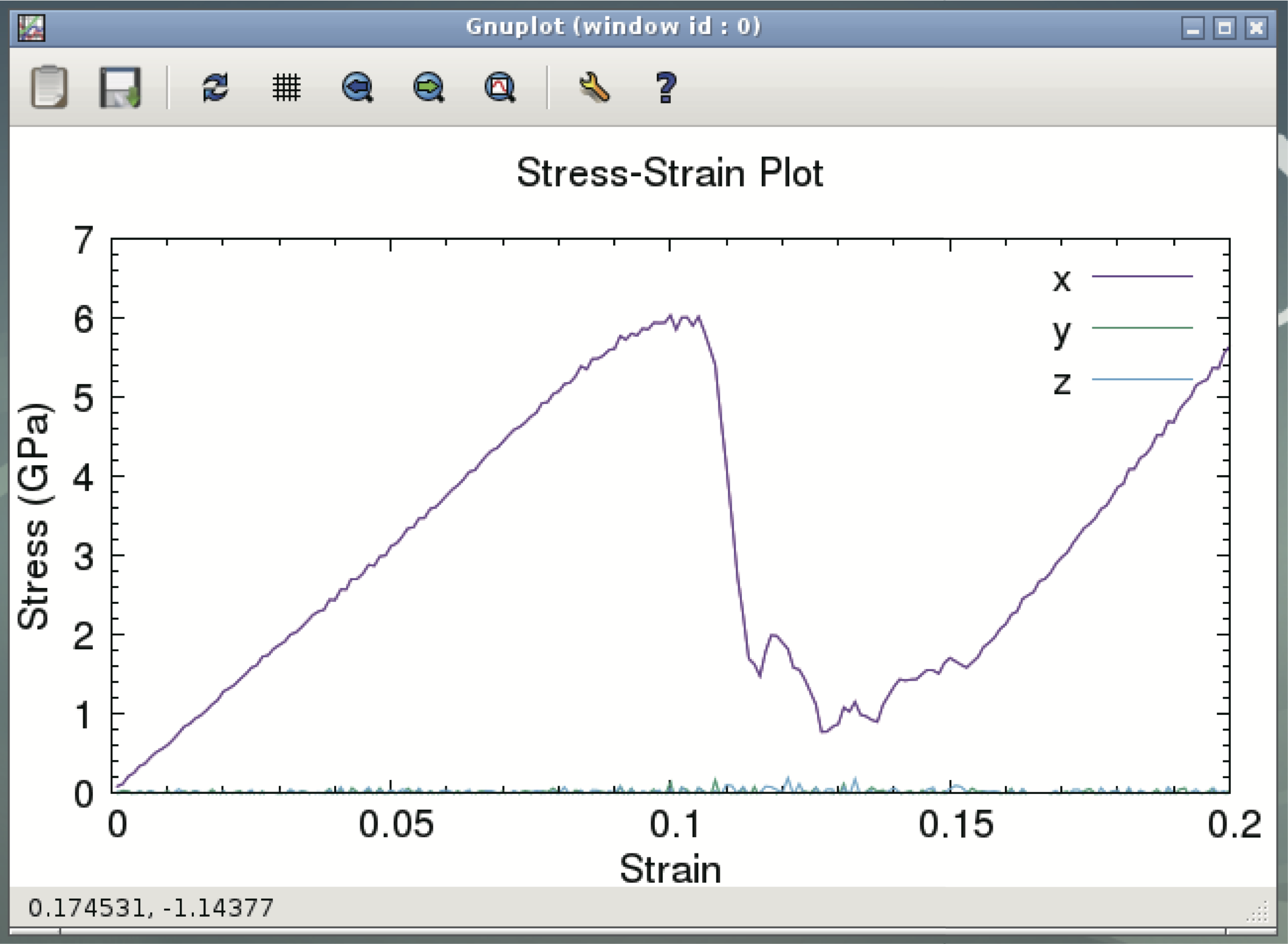
Overview of input files
Let’s take a look at the contents of the input file (in the example above, in.comp.txt) required for LAMMPS calculations (my comments is given in square brackets):
# INITIALIZATION
units metal [Specification of unit system]
dimension 3 [The dimension of the system to handle]
boundary p p p [Periodic boundary condition]
atom_style atomic [Particle type]
variable latparam equal 4.05 [Lattice spacing]
# ATOM DEFINITION
lattice fcc ${latparam} [Face-centered cubic lattice]
region whole block 0 10 0 10 0 10 [Number of unit cells]
[…Omitted…]
# FORCE FIELDS
pair_style eam/alloy [Kind of interatomic potential]
pair_coeff * * Al99.eam.alloy Al [Specification of the force field]
[…Omitted…]
When the unit is specified as metal (the second line), the unit of length is Å. By writing the atom arrangement, the boundary conditions, and the force field specification of the system in the input file, the target and the calculation conditions can be specified in detail (refer to the LAMMPS document for details). The last two lines specify the force field. Here, it is important to select an appropriate force field according to the system to be considered (when using it for research, the user should carefully read the original paper for details of the force field model, and check the properties, accuracy, application limit, etc. of the force field model. For example, in the above example, the Embedded Atom Method (EAM) has been used. In addition to the force field sets prepared in LAMMPS, it is also possible to download and use the force field from the database.
Summary
Since LAMMPS has a lot of setting items, it may seems difficult to get started for the beginners. However, it is not so difficult to find examples of calculation methods and input files on the Web that suit the system that the user want to calculate. If the user is accustomed once, he will notice that LAMMPS is a very powerful software for molecular dynamics.