
Quantum ESPRESSOを用いてGaAsのバンド図を書いてみよう
Last Update:2022/02/12
はじめに
Quantum ESPRESSOはフリーで使える第一原理計算ソフトです。ドキュメントも充実しており、関連ツールも多く公開されています。また平面波基底をつかっているため計算精度を決める制御パラメータが少なく、最初に第一原理計算を試してみるのに最適なソフトの一つといえるでしょう。
本レビューでは、代表的な半導体であるGaAsのバンド計算を行い、バンド図を書くところまで簡単に解説します。環境はMateriApps Live! ver. 3.3を用いました。プレインストールされているQuantum ESPRESSOのバージョンはver. 6.7です。本記事を書くにあたって、下記のチュートリアルを参考にしました。
http://www.cmpt.phys.tohoku.ac.jp/~koretsune/SATL_qe_tutorial/
実行方法
MateriApps LIVE!を起動するまえに、あらかじめVirtualBoxの設定でCPU数をなるべく多くしたほうが計算が早くおわります. 「設定>システム>プロセッサー」とすることでVirtualBox内で使用するCPUプロセッサの数を変更できます(PCのCPUコア数を調べ、それと同じにしておきましょう. 通常はCPU数選択画面で緑のバーの範囲内で最大に設定すればよいはずです.).次に、MateriApps LIVE!上でターミナルを立ち上げておきます(スタートメニューから”System Tools” > “LXTerminal”)。
次にあらかじめ容易されたスクリプトをダウンロードして展開します。
wget https://github.com/cmsi/malive-tutorial/releases/download/tutorial-20210520/qe_GaAs.tgz
tar zxvf qe_GaAs.tgz
cd qe_GaAs電子状態計算を行うには、原子ごとに適切な擬ポテンシャルを使う必要があります。擬ポテンシャルはこのページから探してダウンロードすることができます。ここでは
wget https://pseudopotentials.quantum-espresso.org/upf_files/Ga.pbe-dn-kjpaw_psl.1.0.0.UPF
wget https://pseudopotentials.quantum-espresso.org/upf_files/As.pbe-n-kjpaw_psl.1.0.0.UPFとして、必要な擬ポテンシャルファイルをダウンロードします。バンド計算の実行を行うには、ターミナルから以下のコマンドを実行します。
pw.x < GaAs.scf.in > GaAs.scf.out
pw.x < GaAs.nscf.in > GaAs.nscf.out
bands.x < GaAs.band.in > GaAs.band.out
gnuplot -persistent plot.gp数分で計算が終わるはずです。なお、複数の計算コアが使える場合には、例えば
OMP_NUM_THREADS=1 mpirun -np 2 pw.x < GaAs.scf.inとすれば、スレッド数1、MPI2プロセスの並列計算を行うことができます。このほうが早く計算が終わります。
各コマンドの意味は下記のとおりです。
- pw.xを用いたself-consistent(自己無撞着)計算
- pw.xを用いたnon-self-consistent(非自己無撞着)計算
- bands.xを用いた後処理
- gnuplotを用いたプロット
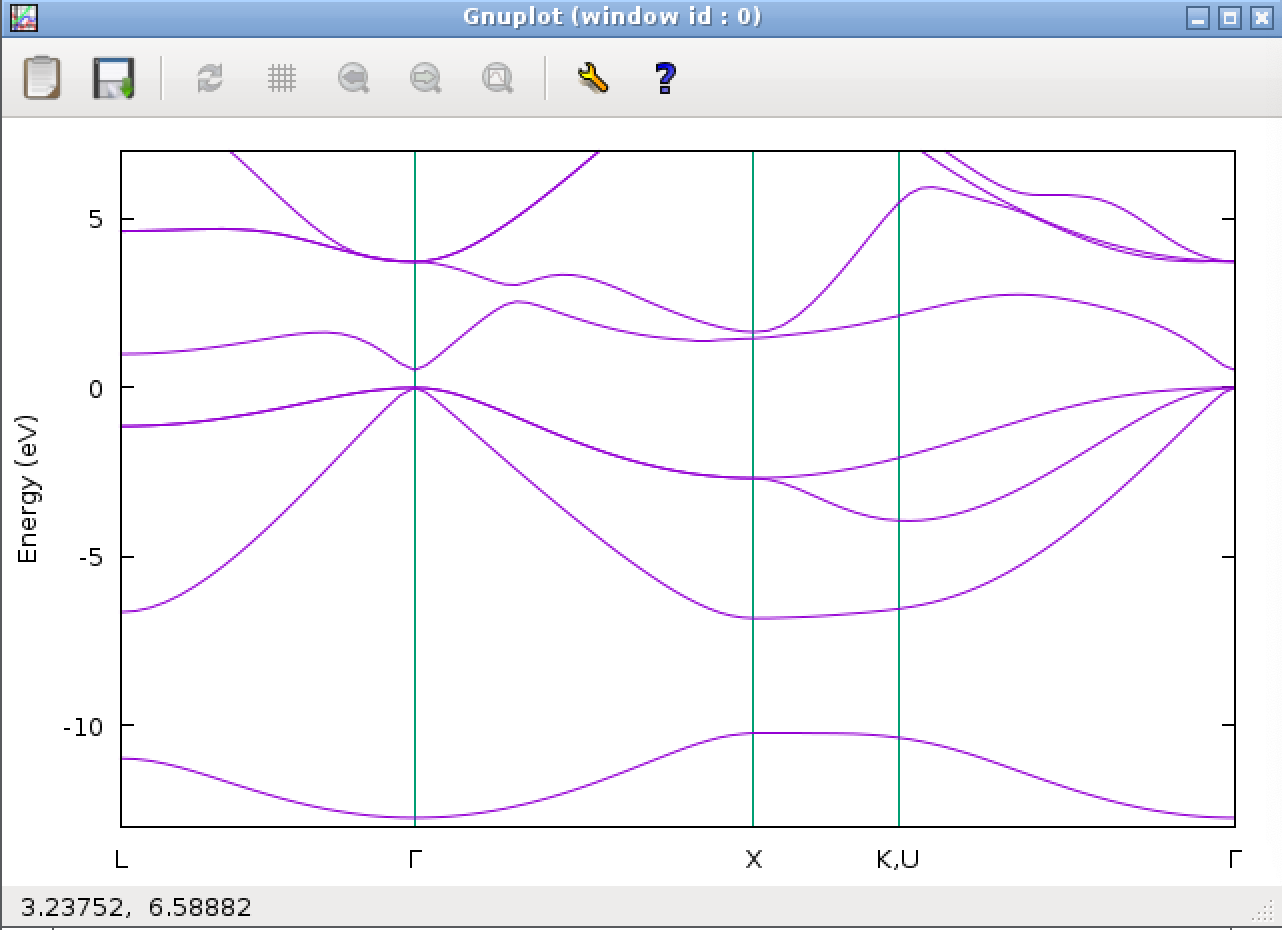
計算には数分かかります。無事計算が終わると、最後に下記のようなバンド図が表示されるはずです。
入力ファイルの解説:自己無撞着計算
self-consistent計算のための入力ファイル(GaAs.scf.in)の中身を詳しくみていきましょう。まずはじめの部分をみてみます。
&control calculation = 'scf' prefix = 'GaAs' pseudo_dir = './' wf_collect = .true. /
&controlから/までがひとまとまりとなっており、その間に”パラメータ名 = 値”の形式でパラメータを設定しています。calculationパラメータは計算モードを指定します。ここでは’scf’とします。これは自己無撞着計算(コーンシャム方程式を自己無撞着に解く)を意味します。prefixパラメータは各種の出力ファイル名のヘッダを指定します。物質名を指定しておくといいでしょう。ここでは’GaAs’とします。pseudo_dirは擬ポテンシャルのファイルがある場所をしていします。あとで後述するように、作業ディレクトリに擬ポテンシャルのファイルを置くことにしますので、今は’./'(同じ場所のディレクトリを意味する)とします。wf_collectは波動関数を保存するオプションです。
&system ibrav = 2 celldm(1) = 10.6867 nat = 2 ntyp = 2 ecutwfc = 60 ecutrho = 244 /
この段落は系の特徴を表す部分です。ibravはブラベー格子の指定の仕方であり、ibrav=2はfcc の単位格子ベクトルを指定します。GaAsの結晶構造はZinc-Blende型結晶構造をとっていますが、これはダイアモンド格子の2つの副格子にGaとAsを配置したもので、ダイアモンド格子の単位格子ベクトルはfccと同じであることに注意しましょう。celldm(1)は格子定数、natはユニットセル内の原子の数、ntypは元素の数をそれぞれ表します。ecutwfc,ecutrhoはそれぞれカットオフエネルギー(単位はリュードベリ)です。カットオフの値を上げるほど精度があがりますが、計算は重くなります。推奨値は擬ポテンシャルの種類によって異なりますが、ここでは後で述べる擬ポテンシャルファイルの冒頭に記述されている推奨値を設定しました。
&electrons mixing_mode = 'plain' mixing_beta = 0.7 conv_thr = 1.0d-8 diago_david_ndim = 4 /
この段落では波動関数の収束に関するパラメータを指定します。conv_thrは収束判定に使うパラメータです。mixing_mode, mixing_betaは収束を早めるためのパラメータになりますが、軽い計算の場合にはあまり気にしなくて構いません。詳しくは公式ページを参照してください。
ATOMIC_SPECIES Ga 69.723 Ga.pbe-dn-kjpaw_psl.1.0.0.UPF As 74.921595 As.pbe-n-kjpaw_psl.1.0.0.UPF
この部分は原子の種類と原子量(本計算では使いません)および擬ポテンシャルファイルを指定します。Quantum ESPRESSOでは擬ポテンシャル法を呼ばれる手法を用いており、内核の電子をとりあつかわず、代わりに内殻電子の価電子への影響を擬ポテンシャルという量で置き換えます。これにより計算量を大きく減らすことができますが、計算結果は擬ポテンシャルの選択によって変わります。よって、擬ポテンシャルは慎重に選ぶ必要があります。すでに述べたように擬ポテンシャルはQuantum ESPRESSOの中のページからダウンロードが可能です。
ATOMIC_POSITIONS Ga 0.00 0.00 0.00 As 0.25 0.25 0.25
これはGaとAsのユニットセル内の内部座標(xyz座標)を表します。
K_POINTS {automatic}
8 8 8 0 0 0
最後の部分で波数ベクトルの点の数(k点)を指定します。最初の3つの数字でx,y,z方向での波数の分割数を指定します。{automatic}の指定があると、自動的に第一ブリルアンゾーン内でk点が離散的にとられます。後ろの3つの数字は波数の原点シフトを記述します。ここではシフトなし(すべて0)としています。k点の数が多いほど正確な計算となりますが、その分計算が遅くなります。自己無撞着計算では、電子密度が収束するまで計算を繰り返すので時間がかかるため、k点の数は少なめにとります。
入力ファイルの解説:非自己無撞着計算
自己無撞着計算で十分正確な電子密度を得ることができますが、バンド図を書くにはk点が足りません。そこで自己無撞着計算で求めた電子密度を元に、求めたい逆格子空間の線分上で多くのk点に対してバンド計算を行います。入力ファイル(GaAs.nscf.in)は自己無撞着計算で用いたファイル(GaAs.scf.in)とほぼ共通です。変更点のみ以下で記述します。まずはじめの部分ですが、
&control calculation = 'bands' prefix = 'GaAs' pseudo_dir = './' wf_collect = .true. /
パラメータcalculationが’bands’に計算されています。これにより自己無撞着計算は行わず、一回だけ状態計算を行います。またsystem部分では、
&system ibrav = 2 celldm(1) = 10.6867 nat = 2 ntyp = 2 ecutwfc = 60 ecutrho = 244 nbnd = 16 /
最後の行でパラメータnbndが追加されています。これによって、計算するバンドの本数が指定されます。最後のk点指定部分は、
K_POINTS {crystal_b}
5
0.00 0.50 0.00 20 !L
0.00 0.00 0.00 30 !G
-0.50 0.00 -0.50 10 !X
-0.375 0.00 -0.675 30 !K,U
0.00 0.00 -1.00 20 !G
と変更されています。{crystal_b}のオプションを指定したときは、逆格子空間中の5つの点(最初の数字で指定)を結んだ4つの線分上のk点を書きます。k点の始点・終点を最初の3つの実数、分割数を4番目に指定します。
後処理
後処理に用いるファイルGaAs.band.inは、短いものです。
&bands prefix = 'GaAs' lsym = .true. /
bands.xのプログラムにこの入力ファイルを渡して実行すると、gnuplot用のプロットファイルbands.out.gnuが生成されます。これをplot.gpファイルで整形しています。
まとめ
Quantum ESPRESSOによるバンド計算を紹介しました。簡単な物質であれば、それほどの手間をかけずにバンド図がかけます。研究の場面だけでなく、大学での授業でも活用していただければと思います。
追記 (2021-05-20)
Quantum ESPRESSO 6.6以降、デフォルトのパラメータに変更があったため、NCSFが収束せず異常終了するようになりました。GaAs.nscf.inのelectronsセクションにdiago_david_ndim = 4を追加した20210520版の入力ファイルを使用するよう記事を修正しました