
QUANTUM ESPRESSOのohtaka(物性研スパコン)でのコンパイル・使用例
Last Update:2024/02/08
#2021/3/19に2020年10月に導入されたohtaka用に書き換えました。
また、moduleやconfigureなどの箇所も追記・修正しました。
コンパイル方法:
ここでは、QUANTUM ESPRESSOの物性研スパコンsystem Bへのインストール方法について紹介します。
まず、ohtaka上にインストール用のディレクトリを作ります。名前はなんでも良いのですが、ここではホームディレクトリ直下にcodeというディレクトリを作ります。
mkdir code
次にcode内にQUANTUM ESPRESSOのレポジトリ(q-e)をcloneして、q-eに移動します。
cd code git clone https://github.com/QEF/q-e.git cd q-e
インストールをするために、まずmoduleを設定します。
module purge module load intel_compiler/2020.4.304 intel_mpi/2020.4.304
configureを利用しコンパイルする準備をします。
./configure --enable-openmp --with-scalapack=intel
OpenMP/MPIハイブリッド並列をしたい場合は––enable-openmpをつけてください。
(ohtakaでは1ノードあたりのコア数が128もあるので、ハイブリッド並列にした方が良いと思われます。)
次にmake と打つとオプションについての結果が出力されます。
to install Quantum ESPRESSO, type at the shell prompt: ./configure [--prefix=] make [-j] target where target identifies one or multiple CORE PACKAGES: pw basic code for scf, structure optimization, MD ph phonon code, Gamma-only and third-order derivatives pwcond ballistic conductance neb code for Nudged Elastic Band method pp postprocessing programs pwall same as "make pw ph pp pwcond neb" cp CP code: Car-Parrinello molecular dynamics tddfpt time dependent dft code gwl GW with Lanczos chains ld1 utilities for pseudopotential generation upf utilities for pseudopotential conversion xspectra X-ray core-hole spectroscopy calculations couple Library interface for coupling to external codes epw Electron-Phonon Coupling with wannier functions gui Graphical User Interface examples fetch from web examples for all core packages test-suite run semi-automated test-suite for regression testing all same as "make pwall cp ld1 upf tddfpt" where target identifies one or multiple THIRD-PARTIES PACKAGES: gipaw NMR and EPR spectra w90 Maximally localised Wannier Functions want Quantum Transport with Wannier functions west Many-body perturbation corrections Without Empty STates yambo electronic excitations with plane waves yambo-devel yambo devel version SternheimerGW calculate GW using Sternheimer equations plumed Metadynamics plugin for pw or cp d3q general third-order code and thermal transport codes where target is one of the following suite operation: doc build documentation links create links to all executables in bin/ tar create a tarball of the source tree tar-gui create a standalone PWgui tarball from the GUI sources tar-qe-modes create a tarball for QE-modes (Emacs major modes for Quantum ESPRESSO) clean remove executables and objects veryclean remove files produced by "configure" as well distclean revert distribution to the original status
今回は基本プログラムである”pw” とその後処理用プログラム”pp” をコンパイルします。
make pw make pp
インストールにかかる時間は約15分です。
無事に終了すると、ディレクトリ“bin” に色々とできます。
以上で、system B上でQUANTUM ESPRESSOを使う準備が完了しました。
実行例:
QUANTUM ESPRESSOのチュートリアルは公式サイト (英語)やこちらのサイト(日本語)が参考になります。
ここではsystem B上での実行例として、チュートリアルサイトにある”シリコンの全エネルギーとバンド構造計算”の練習問題を紹介します。
計算の準備
インプットファイルを用意します。
今回はチュートリアルサイトからダウンロードしたものを利用したいと思います。
ダウンロードしたファイルDay1.tarをホームディレクトリ下に解凍して、以下のコマンドを実行して実行ディクレトリに移動します。
cd ~/Day1/exercise2
また、exercise2/day1_exercise2_instructions.htmlに記載されているように、下記のようなSi.band.inというファイルも作ってください。
Si.band.in
&control
calculation = ‘bands’,
prefix = ‘Si_exc2’,
verbosity = ‘high’
outdir = ‘./tmp/’
pseudo_dir = ‘../pseudo/’
/
&system
ibrav = 2,
celldm(1) = 10.348,
nat = 2,
ntyp = 1,
ecutwfc = 20,
nbnd = 8,
/
&electrons
mixing_beta = 0.7
/ATOMIC_SPECIES
Si 28.086 Si.pbe-rrkj.UPFATOMIC_POSITIONS (alat)
Si 0.0 0.0 0.0
Si 0.25 0.25 0.25K_POINTS (tpiba)
36
0.5 0.5 0.5 1
0.4 0.4 0.4 2
0.3 0.3 0.3 3
0.2 0.2 0.2 4
0.1 0.1 0.1 5
0.0 0.0 0.0 6
0.0 0.0 0.1 7
0.0 0.0 0.2 8
0.0 0.0 0.3 9
0.0 0.0 0.4 10
0.0 0.0 0.5 11
0.0 0.0 0.6 12
0.0 0.0 0.7 13
0.0 0.0 0.8 14
0.0 0.0 0.9 15
0.0 0.0 1.0 16
0.0 0.1 1.0 17
0.0 0.2 1.0 18
0.0 0.3 1.0 19
0.0 0.4 1.0 20
0.0 0.5 1.0 21
0.0 0.6 1.0 22
0.0 0.7 1.0 23
0.0 0.8 1.0 24
0.0 0.9 1.0 25
0.0 1.0 1.0 26
0.0 0.9 0.9 27
0.0 0.8 0.8 28
0.0 0.7 0.7 29
0.0 0.6 0.6 30
0.0 0.5 0.5 31
0.0 0.4 0.4 32
0.0 0.3 0.3 33
0.0 0.2 0.2 34
0.0 0.1 0.1 35
0.0 0.0 0.0 36
最後に実行体をカレントディレクトリにコピーしてきます
cp ~/code/bin/bands.x ./ cp ~/code/bin/pw.x ./ cp ~/code/bin/plotband.x ./
計算の実行
次に、実行のために必要なジョブスクリプト(ohtaka_job.sh)を用意します。
以下のような内容のファイルを作成してください。
#!/bin/sh #SBATCH -J si_ex #SBATCH -p i8cpu #SBATCH -N 1 #SBATCH -t 30:00 #SBATCH --exclusive module purge module load intel_compiler/2020.4.304 intel_mpi/2020.4.304 srun -n 32 -c 4 ./pw.x -nk 32 < Si.scf.in > Si.scf.out srun -n 32 -c 4 ./pw.x -nk 32 < Si.band.in > Si.band.out srun -n 32 -c 4 ./bands.x -nk 32 < bands.in > bands.out srun -n 1 -c 1 ./plotband.x < Si.plotband.in > Si.plotband.out
Si.scf.in、Si.band.in はそれぞれSCF計算およびバンド計算用のインプットファイルです。
続いて、以下のコマンドを実行して計算を行います。
sbatch ohtaka_job.sh
上述のジョブスクリプトを使うと、数十秒ほどで計算が終了します。
実行結果
実行ディレクトリ直下に全エネルギーやバンド構造などの情報が出力されます。
bands.psではバンド構造がSi.band.in内のK_POINTS (tpiba)セクションで指定した通りにプロットされております。(右下図参照。90度回転していると思います。)
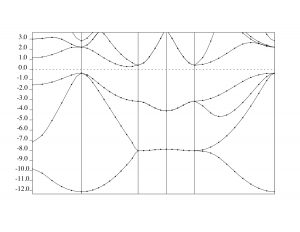
今回のチュートリアルの場合、バンド構造は L (½, ½, ½) →Gamma (0, 0, 0→X (0, 0, 1)→W (0, ½, 1) → X (0, 1, 1)→Gamma (0, 0, 0)というパスで出力されています。