
OpenFermionの使い方
Last Update:2024/12/10
(株式会社アカデメイア)
1.はじめに
OpenFermionは量子ゲートのモデルを用いて量子化学計算を含めたフェルミオン系の計算を実行するためのソフトウェアです。CirqやPySCFなど、外部ソフトウェアと連携するためのプラグインも整備されています。
2.インストール・コンパイル方法(推奨)
ここでは、AnacondaのPython3.6.5、Ubuntu 18.04上でのインストールについて記載します。
インストールはpipコマンドで実行して下さい
$ pip install openfermion
上記コマンド実行で、openfermionがインストールされます。
インストールの確認は以下の通りです。
$ python3
として、起動するPythonの対話モードで、
>>> import openfermion
と入力してEnterを押し、なにもエラーが出なければ、無事にインストール出来ています。
バージョンは
>>> openfermion.__version__
と入力してEnterを押す事で確認が出来、’0.10.0’などのバージョン番号が下の行に出力されます。
3.実行方法
それでは実際にOpenFermionを動かしてみましょう。ここでは、水素分子についての量子化学計算を実行して結合長とそれに対するエネルギーをプロットします。ここの例は、OpenFermionのGitHubリポジトリから抜粋したものです。
https://github.com/quantumlib/OpenFermion/blob/master/examples/openfermion_tutorial.ipynb
(追記:2024年12月現在では上のリンクにアクセスできません。 https://quantumai.google/openfermion/tutorials/intro_to_openfermion に移動したようです。)
from openfermion.hamiltonians import MolecularData
# Set molecule parameters.
basis = 'sto-3g'
multiplicity = 1
bond_length_interval = 0.1
n_points = 25
# Generate molecule at different bond lengths.
hf_energies = []
fci_energies = []
bond_lengths = []
for point in range(3, n_points + 1):
bond_length = bond_length_interval * point
bond_lengths += [bond_length]
description = str(round(bond_length,2))
print(description)
geometry = [('H', (0., 0., 0.)), ('H', (0., 0., bond_length))]
molecule = MolecularData(
geometry, basis, multiplicity, description=description)
# Load data.
molecule.load()
# Print out some results of calculation.
print('\nAt bond length of {} angstrom, molecular hydrogen has:'.format(
bond_length))
print('Hartree-Fock energy of {} Hartree.'.format(molecule.hf_energy))
print('MP2 energy of {} Hartree.'.format(molecule.mp2_energy))
print('FCI energy of {} Hartree.'.format(molecule.fci_energy))
print('Nuclear repulsion energy between protons is {} Hartree.'.format(
molecule.nuclear_repulsion))
for orbital in range(molecule.n_orbitals):
print('Spatial orbital {} has energy of {} Hartree.'.format(
orbital, molecule.orbital_energies[orbital]))
hf_energies += [molecule.hf_energy]
fci_energies += [molecule.fci_energy]
# Plot.
import matplotlib.pyplot as plt
plt.figure(0)
plt.plot(bond_lengths, fci_energies, 'x-')
plt.plot(bond_lengths, hf_energies, 'o-')
plt.ylabel('Energy in Hartree')
plt.xlabel('Bond length in angstrom')
plt.show()
上記ファイルを、h_bond.pyとして保存し、このプログラムを実行します。
python h_bond.py
計算は、5秒程度で終了し、最後にグラフが表示されます。
4.実行結果
計算結果は以下のように標準出力に出力されます。
0.3 At bond length of 0.30000000000000004 angstrom, molecular hydrogen has: Hartree-Fock energy of -0.59382776458494 Hartree. MP2 energy of -0.5997818888874376 Hartree. FCI energy of -0.6018037168352988 Hartree. Nuclear repulsion energy between protons is 1.7639240286333335 Hartree. Spatial orbital 0 has energy of -0.8028666187118976 Hartree. Spatial orbital 1 has energy of 1.368938795250223 Hartree. 0.4 At bond length of 0.4 angstrom, molecular hydrogen has: Hartree-Fock energy of -0.904361397713845 Hartree. MP2 energy of -0.9114367296997896 Hartree. FCI energy of -0.9141497082141279 Hartree. Nuclear repulsion energy between protons is 1.322943021475 Hartree. Spatial orbital 0 has energy of -0.7452125332909346 Hartree. Spatial orbital 1 has energy of 1.167416395038123 Hartree. (中略) 2.5 At bond length of 2.5 angstrom, molecular hydrogen has: Hartree-Fock energy of -0.7029435983728299 Hartree. MP2 energy of -0.8535394695489013 Hartree. FCI energy of -0.9360549198442759 Hartree. Nuclear repulsion energy between protons is 0.21167088343600005 Hartree. Spatial orbital 0 has energy of -0.2144671917818725 Hartree. Spatial orbital 1 has energy of 0.04995242289757026 Hartree.
上記では、ボンド長と、それに対応した分子のエネルギー(Hartree-Fockエネルギー、FCI(Full-CI)エネルギー、軌道のエネルギー等)の情報が出力されます。また、計算後、水素分子の結合長に対するHartree-FockエネルギーとFull-CIエネルギーが以下のようにプロットされます。
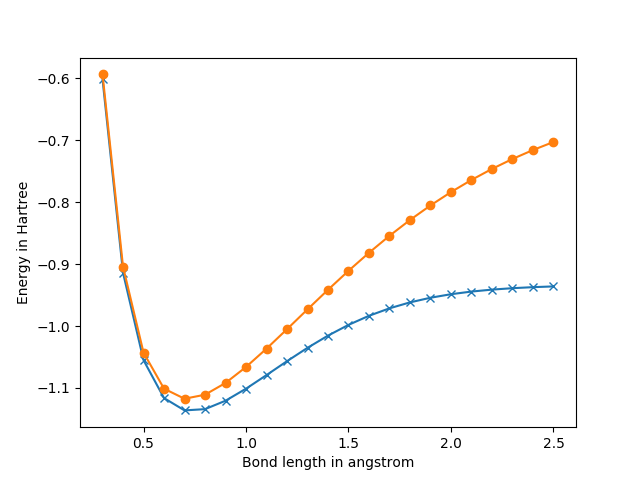
上記図で、●の点がHartree-Fockのエネルギー、×の点がFull-CIのエネルギーとなります。
5.終わりに
ここではOpenFermionを用いた簡単な計算例を紹介しました。量子ゲート模型を用いて量子化学等のフェルミオン系の計算を容易に行う事が可能です。この紹介例の他にもGithubリポジトリのexamplesにJupyter Notebook形式で例がありますので興味のある方はご確認いただければと思います。
https://github.com/quantumlib/OpenFermion/blob/master/examples
(追記:2024年12月現在では上のリンクにアクセスできません。https://quantumai.google/openfermion/overview に移動したようです。)