
PySCFを使った水素分子H2の計算例
Last Update:2024/12/10
はじめに
PySCFは様々な量子化学手法が実装されているPythonパッケージです。ドキュメントも充実しており、インストールや使い方が簡便なため、量子化学分野に関わってこなかった方でも比較的簡単に実行できるようになっています。
ここでは物性研スパコン(system B)上で、PySCFによる水素分子の解離エネルギーの計算例を紹介します。なお筆者は量子化学の計算手法に関しては聞いたことがある程度なものの、実際の計算は未経験です。今回の記事で試してみる手法はRHF(制限Hartree-Fock)、MP2(Møller-Plesset 二次摂動)、CCSD(二電子励起まで含めたカップルド・クラスター法)、CASSCF(完全活性空間 自己無撞着場法)、Full CI(完全 配置間相互作用法)です。なお、物性研でなくPythonが使える環境であれば自分のパソコンやクラスター上でも動くと思いますので、物性研スパコンが使えない方も是非お試しください。
インストール方法
はじめに、PySCFをインストールしましょう。公式サイトに色々な方法が記載されていますが、今回は一番簡単そうなpipコマンドを使ったインストールを試します。
まず、物性研スパコン(system B)にログインした後、以下のコマンドを実行してPythonの環境を整えます。
source /home/issp/materiapps/tool/env.sh続いて、pipコマンドを実行します。
pip install --user pyscfuserオプションをつけると、自分のHOMEディレクトリ下の.local/libにpyscfがインストールされます。これでインストール作業は終了です。
実行方法
それでは、実際にPySCFを動かしてみたいと思います。HOMEディレクトリ下に実行用のディレクトリを作成して移動します。
mkdir pyscf_h2mol
cd pyscf_h2mol続いて、実行用のPythonスクリプトを用意します。wgetを使ってスクリプトh2mol.pyをダウンロードして、テストディレクトリpyscf_h2molに持ってきてください。公式ドキュメントを参考に作成しているので、スクリプトの説明はそちらをご参照ください。
wget https://ma.issp.u-tokyo.ac.jp/wp-content/uploads/sites/3/2019/04/h2mol.py最後に下記のコマンドを打つと、先ほどダウンロードしたスクリプトを用いた計算が実行されます。
python h2mol.py > logだいたい数十秒程度でプログラムが終了すると思います。
実行結果
スクリプトを実行するとresults.datに結果が出力されます。これをコマンドライン上で
head -2 results.datと打つと
#r[Bohr] Energy[a.u] (RHF, CASSCF, MP2, CCSD, Full CI)
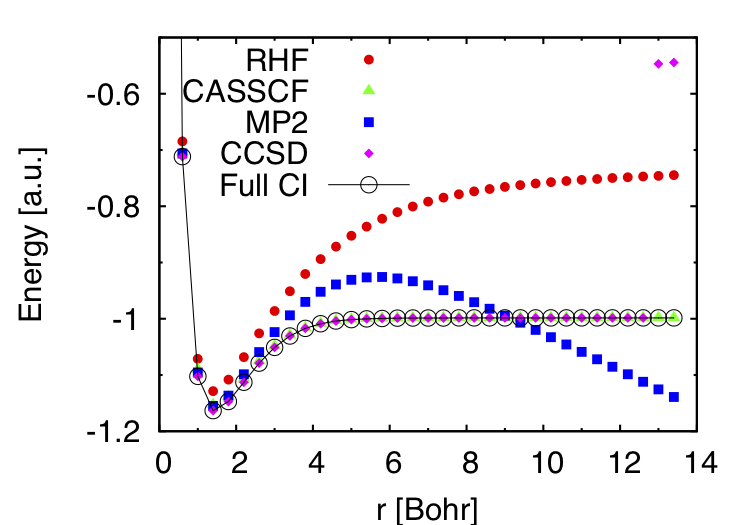
0.2000000000 2.3747139119 2.3584271235 2.3555487509 2.3509944253 2.3509944329と出力されます。一列目が水素原子間の距離を、それ以外の列は各計算手法によって得られたエネルギーです。順番は一行目に書いてある通りです。gnuplotでプロットしてみると下の図のようになります。白抜きの完全CIの結果が厳密解です。平均場近似であるRHFやそこからの二次摂動であるMP2だと解離エネルギーをうまく表現できていないことがよくわかります。一方、遠方の原子間距離でCCSDの結果が高エネルギー側で収束してしまっていますが、CASSCFとCCSDは厳密解をよく再現できています。
なおスクリプトの10行目を
mol = gto.M(atom='H 0 0 0; Li 0 0 '+ str(z), basis='ccpvdz', unit='Bohr')
と書き換えるとLiH分子の計算になります。そうするとCASSCFとCCSDの結果に違いが出てきますので、余裕があったら試してみてください。
終わりに
ここではPySCFを用いた水素分子計算の例を紹介しました。PySCFは今回試した計算手法以外にもUHFやDMRGプログラムのBLOCKとのインターフェイスなども備えてあります。また、分子系以外にも周期系への適用もできる柔軟なパッケージになっているので、これを機に是非利用してみてください!
#2022/3/11: リンクやスクリプト、本文を微修正しました。