
Building a material surface model by ASE (Atomic Simulation Environment)
Last Update:2025/06/11
Introduction
Here, I explain how we can build a material surface model (slab model) by ASE(Atomic Simulation Environment).
Preparation of bulk structure data (cif file)
At first, we download a bulk structure data from Materials Project(https://materialsproject.org/). When you use Materials Project, You need to login. However, you can login the site by using some general account such as gmail.
Here, let us try to build Ag(111) surface as an example.
Click “App” button of the above menu tab, and choose “Materials Explorer”.
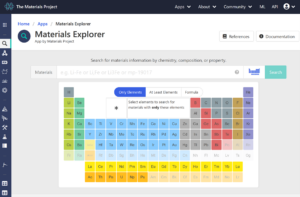
After clicking “Ag”, click “search”. Then, some candidates appear.
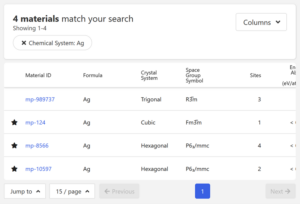
In this review, I choose ID mp-124.
Download a cif file by clicking “Export as” button in the upper right corner of the structure figure.
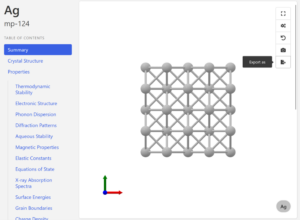
Build a surface structure (slab structure)
ASE has Python interface. Next, let’s write a code of Python to read cif file of the primitive cell, build a surface structure, and export the cif file.
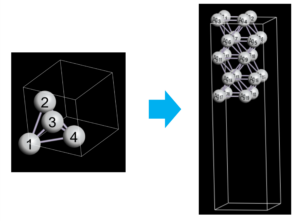
At first, let’s build Ag(111) surface with 5 atomic layers, and insert 10 Å vacuum region to avoid interaction between two surfaces due to the periodicity.
The sample code is shown below.
'''
Surface builder by using ASE (https://wiki.fysik.dtu.dk/ase/index.html)
* Installation
https://wiki.fysik.dtu.dk/ase/install.html
* Usage
Prepare a ciffile of a primitive cell
and information of a surface Miller index and the number of layers.
Execute the following:
$ python3 surface_builder_ase.py input.cif output.cif
'''
import os
import sys
import numpy as np
def edit_structure_ase(filename_input,filename_output):
### Reference:
### https://wiki.fysik.dtu.dk/ase/ase/atoms.html
### https://wiki.fysik.dtu.dk/ase/ase/build/surface.html?highlight=ase%20build%20surface#ase.build.surface
### https://wiki.fysik.dtu.dk/ase/ase/build/tools.html
from ase import io
from ase import Atoms
from ase.build import surface, cut, add_vacuum
#####################
### read cif file ###
#####################
atoms_input = io.read(filename=filename_input)
#####################
### build surface ###
#####################
indices = (1,1,1) # Miller index
layers = 5 # number of layers
vacuum = 10.0 # vacuum thickness
### ase.build.surface(lattice, indices, layers, vacuum=None(Ang), tol=1e-10, periodic=False)
atoms = surface(atoms_input, indices, layers, vacuum)
### ###
######################
### write cif file ###
######################
io.write(filename=filename_output, images=atoms)
return
if __name__ == '__main__':
filename_input=sys.argv[1]
filename_output=sys.argv[2]
filepath = os.getcwd()
current_dir = os.path.basename(filepath)
edit_structure_ase(filename_input,filename_output)
Reading and writing cif files can be performed by “ase.io.read” and “ase.io.write”. The surface structure is built by “ase.build.surface”. The direction of the surface, the number of layers and the width of vacuum region are specified in the part just before “ase.build.surface”.
The above python code can be performed by
$ python3 surface_builder_ase.py input.cif output.cifThe produced cif file of Ag(111) surface structure can be visualized easily by OpenMX viewer or VESTA.
The following figures show the original primitive cell (left) and the generated surface structure (right) visualized by OpenMX Viewer.
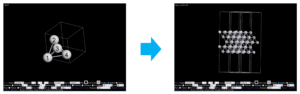
※ You can also specify the cutting surface by a surface over which the lattice vectors a and b stretch. In that case, “ase.build.cut” is useful. The sample code is shown below.
'''
Surface builder by using ASE (https://wiki.fysik.dtu.dk/ase/index.html)
* Installation
https://wiki.fysik.dtu.dk/ase/install.html
* Usage
Prepare a ciffile of a primitive cell
and information of a surface Miller index and the number of layers.
Execute the following:
$ python3 surface_builder_ase.py input.cif output.cif
'''
import os
import sys
import numpy as np
def edit_structure_ase(filename_input,filename_output):
### Reference:
### https://wiki.fysik.dtu.dk/ase/ase/atoms.html
### https://wiki.fysik.dtu.dk/ase/ase/build/surface.html?highlight=ase%20build%20surface#ase.build.surface
### https://wiki.fysik.dtu.dk/ase/ase/build/tools.html
from ase import io
from ase import Atoms
from ase.build import surface, cut, add_vacuum
from ase.build import root_surface, root_surface_analysis
#####################
### read cif file ###
#####################
atoms_input = io.read(filename=filename_input)
#####################
### build surface ###
#####################
### ase.build.surface(lattice, indices, layers, vacuum=None(Ang), tol=1e-10, periodic=False)
#atoms = surface(atoms_input, (1, 1, 1), 5, vacuum=10.0) # system, Miller index, number of layers
#atoms = atoms_input
### ###
################
### cut cell ###
################
###ase.build.cut(atoms, a=(1, 0, 0), b=(0, 1, 0), c=None, clength=None, origo=(0, 0, 0), nlayers=None, extend=1.0, tolerance=0.01, maxatoms=None)
atoms = cut(atoms_input, a=(1, 1, 0), b=(-1, 1, 0), nlayers=5)
add_vacuum(atoms, vacuum=10.0)
######################
### write cif file ###
######################
io.write(filename=filename_output, images=atoms)
return
if __name__ == '__main__':
filename_input=sys.argv[1]
filename_output=sys.argv[2]
filepath = os.getcwd()
current_dir = os.path.basename(filepath)
edit_structure_ase(filename_input,filename_output)
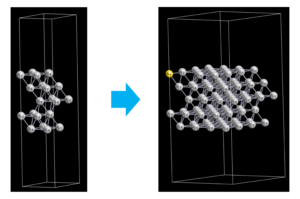
Supercell, Replace, Remove
In ASE, the Atoms object provides the methods such as building supercells (repeat), replacing atoms (get_chemical_symbols, set_chemical_symbols) and removing atoms (pop).
In the example code below, the Ag(111) surface is extended to the 2×2 supercell. In addition, the first Ag atom is replaced to Au, and the second atom is removed.
'''
Surface builder by using ASE (https://wiki.fysik.dtu.dk/ase/index.html)
* Installation
https://wiki.fysik.dtu.dk/ase/install.html
* Usage
Prepare a ciffile of a primitive cell
and information of a surface Miller index and the number of layers.
Execute the following:
$ python3 surface_builder_ase.py input.cif output.cif
'''
import os
import sys
import numpy as np
def edit_structure_ase(filename_input,filename_output):
### Reference:
### https://wiki.fysik.dtu.dk/ase/ase/atoms.html
### https://wiki.fysik.dtu.dk/ase/ase/build/surface.html?highlight=ase%20build%20surface#ase.build.surface
### https://wiki.fysik.dtu.dk/ase/ase/build/tools.html
from ase import io
from ase import Atoms
from ase.build import surface, cut, add_vacuum
#####################
### read cif file ###
#####################
atoms_input = io.read(filename=filename_input)
#####################
### build surface ###
#####################
indices = (1,1,1) # Miller index
layers = 5 # number of layers
vacuum = 10.0 # vacuum thickness
### ase.build.surface(lattice, indices, layers, vacuum=None(Ang), tol=1e-10, periodic=False)
atoms = surface(atoms_input, indices, layers, vacuum)
### ###
#################
### supercell ###
#################
atoms = atoms.repeat((2,2,1))
##############################
### replace atomic species ###
##############################
list_chemical_symbols = atoms.get_chemical_symbols()
#print("list_chemical_symbols:")
#print(list_chemical_symbols)
list_replace_atoms = [] # index starts from 1
list_replace_atoms = [[1,"Au"]]
#list_replace_atoms = [[3,"Au"],
# [4,"Au"]]
for index in list_replace_atoms:
list_chemical_symbols[index[0]-1] = index[1]
atoms.set_chemical_symbols(list_chemical_symbols)
#print("list_chemical_symbols:")
#print(list_chemical_symbols)
### ###
####################
### remove atoms ###
####################
list_remove_atoms = [2] # index starts from 1
#list_remove_atoms = [1] # index starts from 1
for i, index_atom in enumerate(list_remove_atoms):
atoms.pop(index_atom-i-1)
######################
### write cif file ###
######################
io.write(filename=filename_output, images=atoms)
return
if __name__ == '__main__':
filename_input=sys.argv[1]
filename_output=sys.argv[2]
filepath = os.getcwd()
current_dir = os.path.basename(filepath)
edit_structure_ase(filename_input,filename_output)
Sometimes a “warning” message appears, but cif file is successfully generated. When you actually build such a structure, it is useful to visualize the structure by structure viewers such as VESTA and OpenMX Viewer. These viewers make it easy to check the index of atoms which is replaced or removed.
Summary
In this review, I explained how we can build surface (slab) structures. In ASE, there are a lot of methods and tools which I have not introduced here. You can find other methods and tools in the ASE official site.