
Experience of LAMMPS simulations on MateriApps LIVE!(2019.7.11)
Last Update:2021/12/09
I performed an MD simulation by using LAMMPS(11 Aug 2017) on MateriApps LIVE!.
Preparation:
- See Wiki of MaterApps LIVE! for details on how to use MateriApps LIVE! .
- The samples for LAMMPS are in the directory “/usr/share/lammps/examples/melt” on MaterialApps LIVE!
$ cp -r /usr/share/lammps/examples/melt ./ $ cd melt
- Install VMD to plot output of LAMMPS by launching the setup tool on the command line.
$ vmd-setup
After inserting your username and password, the installation of VMC will start.
If you have not used VMD before, please register to download VMD at the official site. You do not need to download VMD from the official site, as vmd-setup will handle that for you. You can thus close the browser after creating your account for download.
Run LAMMPS:
- Run the following command.
$ lammps -in in.melt
In this sample, Lennard-Jones (LJ) particles are arranged in a FCC lattice.
- Next, the input file, in.melt, is modified to output a visualization file. Execute the following command and copy the input, in.melt.
$ cp in.melt in.melt_dump
I get in.melt_dump, and after opening in.melt_dump by editor (vi, emacs, etc.), delete “#” in “#dump id all atom 50 dump.melt” in this file.
- Run the modified in.melt_dump.
$ lammps -in in.melt_dump
- The output dump.melt can be displayed using the visualization tool VMD. First, open VMD by
$ vmd
From “File => New Molecule”, launch “Molecule File Browser” and “Molecule File Browser” will open. After that, click “Browse… “ to select the file “dump.melt” and set “LAMMPS Trajectory” in “Determine file type”, and click “Load”. I think you see very noisy trajectory of LJ particles.

To enhance visibility, click “Graphics=>Representations” in “VMD Main” and “Graphical Representations” window will open. After I change “Lines” to “VDW” in “Drawing Method” and set 0.2 in “Sphere Scale”, the following figure can be obtained.
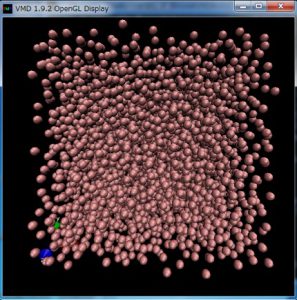
Back to “VMD Main” and click “Play forward” (right bottom button), you will see MD simulations in melting process of FCC lattice of LJ particles.