
Simulation of Magnetism and Self-Assembled Nanostructures in Magnetic Semiconductors using AkaiKKR
Kazunori Sato (Division of Materials and Manufacturing Science, Graduate School of Engineering, Osaka University)
Magnetic semiconductors are substances created by heavily doping of magnetic impurities (such as manganese (Mn)) into semiconductors such as gallium arsenide (GaAs) and indium arsenide (InAs). They exhibit ferromagnetism, and as ferromagnetism can be controlled by modulating the carrier density, they are being studied as promising candidates for semiconductor spintronics. Because impurities are randomly distributed in the crystal, conventional band calculations that assumes the translational symmetry are not applicable to magnetic semiconductors. The coherent potential approximation (CPA) method is used for treating such systems without translational symmetry, and when combined with Korringa–Kohn–Rostoker (KKR) method, which is a band calculation method that utilizes Green’s function, it can calculate the electronic structure of disordered alloys. The open software for this combined method (KKR–CPA method) has been developed as AkaiKKR. As a typical example of KKR-CPA method, the author’s research group made predictions on the magnetic properties of magnetic semiconductors. Our studies have shown that the estimated Curie temperatures agree reasonably with experiments for typical magnetic semiconductors (Fig. 1) [1]. In this calculation, the effective exchange interactions between Mn within the GaAs crystal are calculated based on the KKR-CPA, and the Curie temperatures are estimated using the Monte Carlo method. KKR method can calculate system’s Green’s function and it has been applied for various systems such as disordered systems and systems containing impurities and/or defects. KKR method can also calculate electron transport phenomenon and can be employed as Order-N calculations (also known as the screened KKR method). Recent experiments in magnetic semiconductors have shown that magnetic impurities in various magnetic semiconductors separate and they form self-organized nanostructures. The simulation can be applied to such complex phenomena in real materials (Fig. 2), and the technique has been applied for the interpretation of experimental research and for designing materials with new functions [2, 3]. The calculations introduced here were carried out using a standard computer cluster.
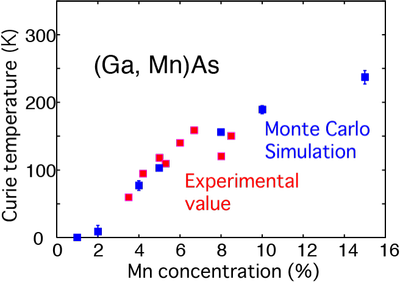
Figure 1: Comparison between experimental (red squares) and calculated (blue squares) values for Curie temperature of Mn-doped GaAs. Calculation was based on Monte Carlo method employing exchange interactions estimated by KKR–CPA method (reference paper: [1]).

Figure 2: Simulation of self-assembled nanostructures of Cr impurities in Cr-doped ZnTe (reference paper: [2])
Reference:
[1] K. Sato, L. Bergqvist, J. Kudrnovský, P. H. Dederichs, O. Eriksson, I. Turek, B. Sanyal, G. Bouzerar, H. Katayama-Yoshida, V. A. Dinh, T. Fukushima, H. Kizaki, and R. Zeller, Rev. Mod. Phys. 82, 1633 (2010).
[2] T. Dietl, K. Sato, T. Fukushima, A. Bonanni, M. Jamet, A. Barski, S. Kuroda, M. Tanaka, Pham Nam Hai, and H. Katayama-Yoshida, Rev. Mod. Phys. 87, 1311 (2015).
[3] T. Fukushima, K. Sato, H. Katayama-Yoshida and P. H. Dederichs, Jpn. J. Appl. Phys. 45, L416 (2006)