
K-path generation using SeeK-path for drawing a band structure
Last Update:2025/08/29
1. Abstract
When you perform electronic state calculations to create a band diagram, you must specify high-symmetry points in the wavenumber space and a wavenumber path connecting them. In principle, high-symmetry points can be calculated manually from the primitive lattice vectors and reciprocal lattice vectors of a crystal, but considering wavenumber paths in three dimensions is tedious and time-consuming. By using SeeK-path, you can easily grasp the high-symmetry point visually, and in addition, it is possible to automatically generate the wavenumber specification part of the input file of first-principles calculation software. In this review, we obtain a crystal structure file (cif file) of semiconductor GaAs from the Materials Project database and generate a wavepath using the SeeK-path web interface.

2. Obtaining a crystal structure
Launch a browser and go to the GaAs page in Materials Project. You will need to log in to the Materials Project to get the crystal structure file, but you can do so quickly by using your Gmail account, for example. Once logged in, click “Export as” botton in the red circle below and select “CIF (Symmetrized)” to download the crystal structure file.

3. How to use the Web interface of SeeK-path
From your browser, enter the SeeK-path web interface . Press “Select File” button and select the CIF file you just downloaded (GaAs.cif). Select “CIF File (.cif) [parser: pymatgen]” for the file format immediately below.
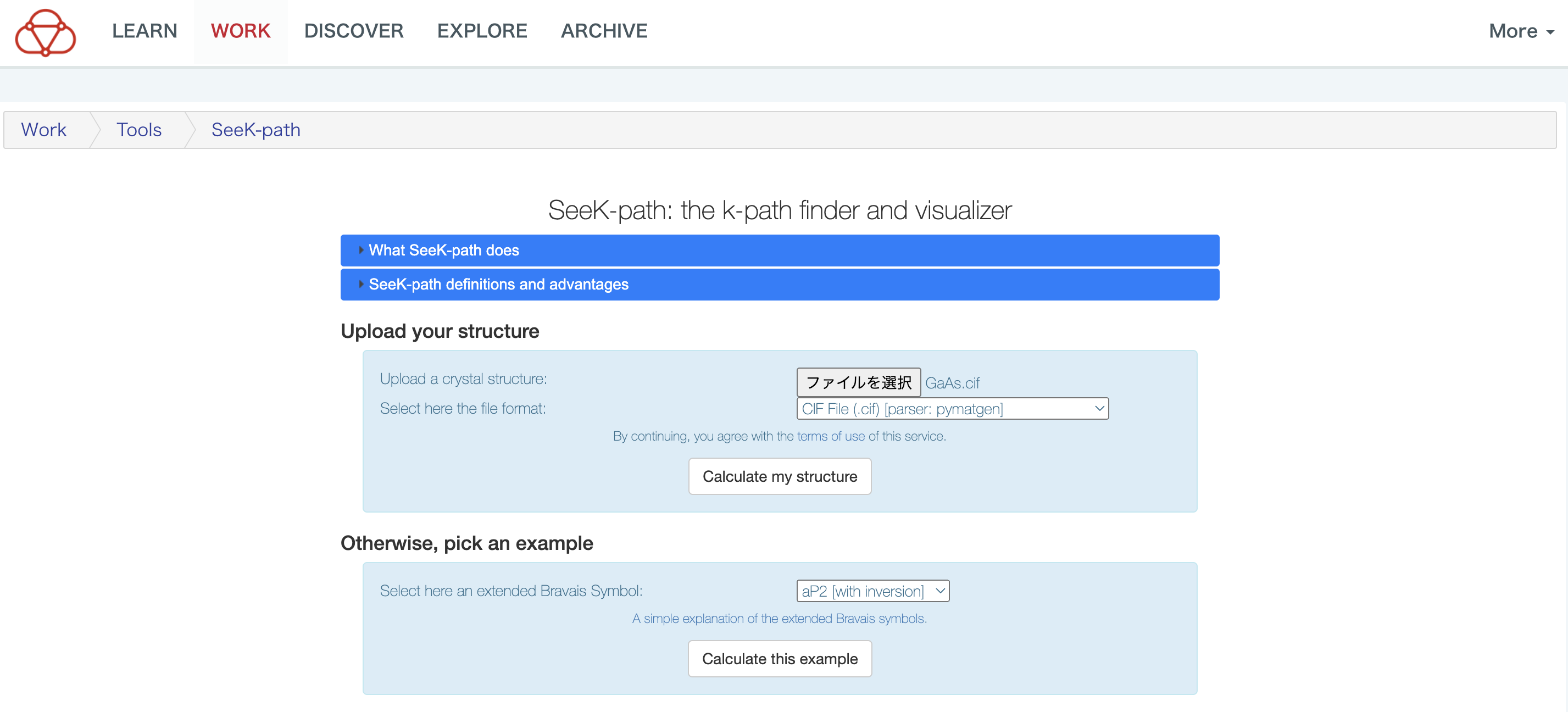
Then press “Calculate my structure” button below it. After a few moments, a diagram of the reciprocal space (the first Brillouin zone) and a diagram of the unit cell appear. Symbols such as K, L, U represent high symmetry points in wavenumber space. You can reorient the two graphs by double-clicking and then clicking and dragging the mouse.

Below it is information about the reciprocal lattice vector and the high symmetry points in the wavenumber space, as well as information about the atomic positions in the unit cell.

Files that can be used as part of the input file of first-principles calculation software are displayed below. For example, if you open the “Quantum ESPRESSO pw.x input” tab, you will see the text available as part of the input file:
