
Calculation of a spectral function of the impurity Anderson model using flexible DM-NRG
Last Update:2021/12/09
Introduction
flexible DM-NRG is an open-source program that solves impurity models by Numerical Renormalization Group (NRG). To get the source code, fill out the form on the licensing Web page and submit it. After a while, you will receive the URL of the source code. The license of flexible DM-NRG is GNU GPL. The file name of the code I got is flexible-dmnrg-1.0.0.tgz. In the following, assuming that this source code is saved in the home directory of MateriApps LIVE! In this tutorial, let’s compile the program and calculate the spectral function of the impurity Anderson model. The version of MateriApps LIVE! Used is 2.4.
How to install
First, unzip the source code and go down to the newly created directory:
$ tar zxvf flexible-dmnrg-1.0.0.tgz $ cd flexible-dmnrg-1.0.0
Next, download and unzip the script file to compile and run:
$ wget https://github.com/cmsi/malive-tutorial/releases/download/tutorial-20200202/flexible_dmnrg.tgz $ tar zxvf flexible_dmnrg.tgz
Compile with the following command:
$ sh compile.sh
If the code is compiled without problems, you should have executable files such as fnrg and sfb in the same directory.
How to run
Once the installation is complete, let’s run it immediately. Basically, prepare a file named “input.dat” and execute the program fnrg. To calculate the spectral function, the program sfb is subsequently executed. The format of “input.dat” is generally complex. This is to accommodate various models. However, for a simple model, templates are provided in the directory input_files, so copy it and rename it to “input.dat”. In “input.dat”, in addition to physical parameters, it is possible to specify the number of states to be left in the renormalization step and the energy renormalization coefficient Λ.
Here, let’s execute the calculation of the numerical renormalization group for the impurity Anderson model using a script prepared in advance. Enter the following command:
$ sh run.sh
As a result, NRG calculation is performed changing interaction parameters U, each of which a spectral function is calculated. The output files named “spectral_func_U _ ****.dat” are generated in the same directory (*** indicates a value of U). To display them, use the following command.
$ gnuplot -persistent plot.gp
If executed successfully, you should get a graph shown below.
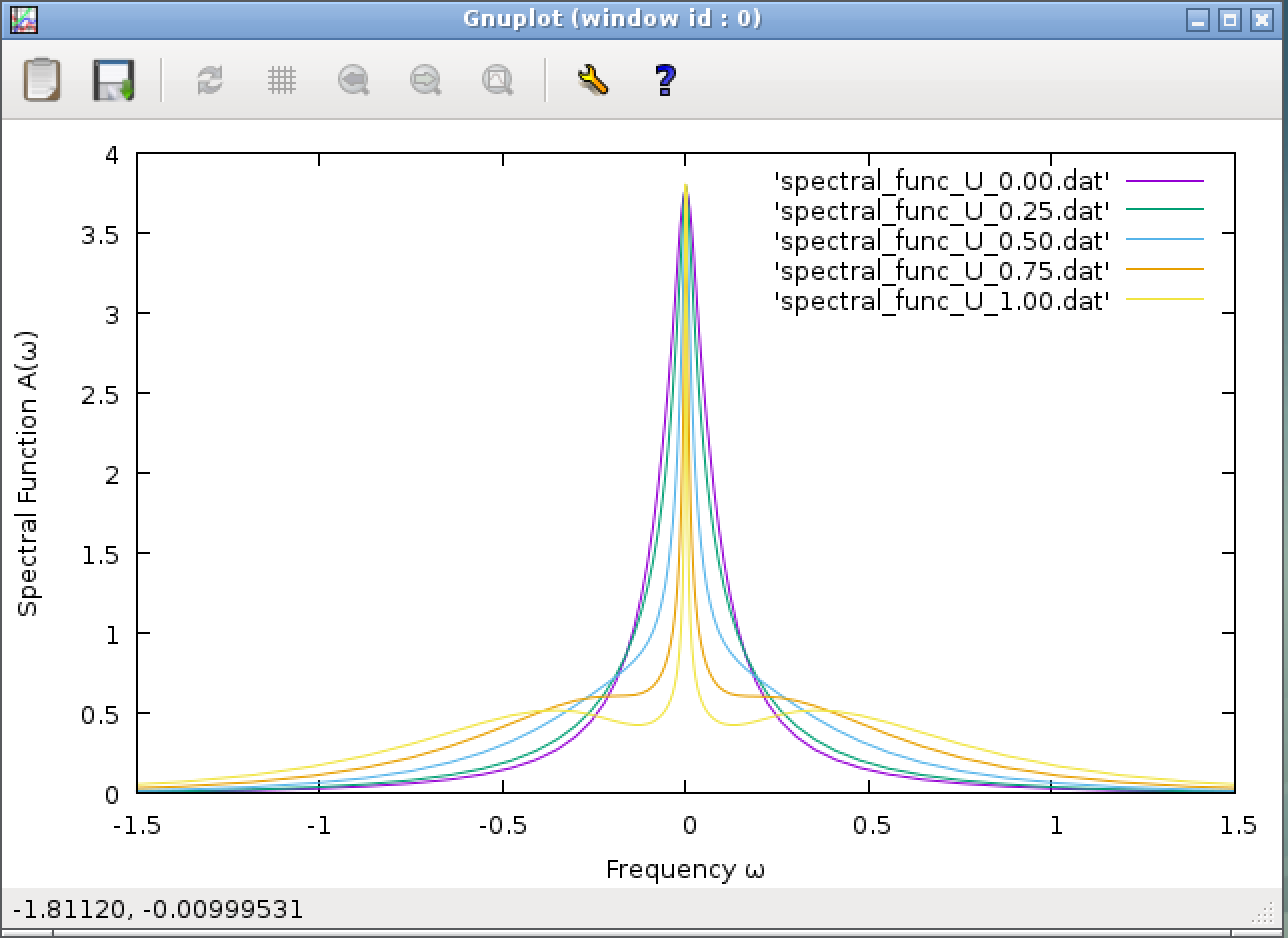
In the Anderson model, in addition to the magnitude of the interaction U, the width of the spectrum function at U = 0 is an important parameter. Actually, the spectrum function when U=0 is written as A(ω) = (Γ/π)/(ω2+Γ2). In this calculation, the hopping between the impurity and the conduction electron chain is set to t = 0.25 (the line “t = 0.25” can be found in the “input.dat” file), and the line width is calculated from this hopping as Γ=πt2/2 (here Γ = 0.098). The spectral function of U = 0 calculated from this and the result of U = 0 in the NRG calculation are slightly different, but this is because the renormalization coefficient Λ (here, 2) is too large, or because the number of states to be left (here, 100) is too small. Adjusting these parameters improves the match (though the calculation becomes heavy).
Summary
The flexible DM-NRG manual provides detailed information, but is not user-friendly. It may take some time to find how to control the parameters and the output files until you check the first calculation example. I hope this tutorial will help the beginners. Once the calculations have been made, it is easier than expected to calculate other parameters and models while referring to the manual. I hope that you will enjoy to use flexible DM-NRG.