
Large-Scale Molecular Structure Calculation Based on MP2 Method (Wave Functional Theory that Includes Electron Correlation) with DC
One of the most commonly used methods for calculating the electronic state of molecules and solids is the density functional theory (DFT). DFT is an excellent method that can provide a good balance between computational cost and accuracy. However, the calculation results largely depend on the “functional” used in the computation, and there does not exist a functional that is absolutely optimal for all models. In the field of quantum chemistry, there are traditional and systematic methods known as the wave function theory, which offers a clear strategy for improving the accuracy of the theory. However, their computational cost is huge compared to DFT, especially for large molecules, even when using the MP2 method that includes minimum electron correlation. Furthermore, the additional cost required for the calculation of the energy gradient for determining the molecular structure is also much greater than that required with DFT, making calculations of large-scale systems difficult.
To enable the calculation of electronic state of large-scale molecular systems using the wave function theory, we have utilized the divide-and-conquer (DC) theory that dramatically reduces computational time, and we developed a computer program “DC” that takes advantage of this theory. Refer to this page (in Japanese) for an overview of the theory.
The figure shows the computational time (above) and memory use (below) required for calculating the DC-MP2 energy gradient of a model molecule, polyene (CnHn+2) [1]. Using normal methods, a larger molecular size (along the horizontal axis) leads to a dramatic increase in the computational time and memory use. If DC is used, the computational time only increases linearly, and memory is kept constant. This calculation was carried out on a single-node PC, but DC-MP2 has also been developed for two-stage hierarchical parallel calculation [2], and has been successfully run using the K supercomputer. Wave function theory methods such as MP2 are critically important for the analysis of biopolymers, as they can incorporate dispersion ab initio, unlike DFT that can only handle it empirically. They have therefore been applied to the calculation of proteins [3-5].
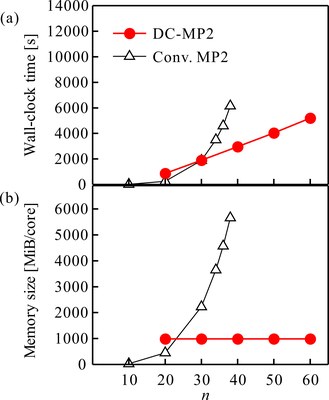
Figure: (a) Computational time and (b) Memory use required for the calculation of DC-MP2 energy gradient of polyene CnHn+2
[1] M. Kobayashi and H. Nakai, J. Chem. Phys. 138, 044102 (2013).
[2] M. Katouda, M. Kobayashi, H. Nakai, and S. Nagase, J. Comput. Chem. 32, 2756 (2011).
[3] P. Saparpakorn, M. Kobayashi, S. Hannongbua, and H. Nakai, Int. J. Quantum Chem. 113, 510 (2013).
[4] P. Saparpakorn, M. Kobayashi, and H. Nakai, Bull. Chem. Soc. Jpn. 86, 67 (2013).
[5] T. Yoshikawa, M. Kobayashi, A. Fujii, and H. Nakai, J. Phys. Chem. B 117, 5565 (2013).